Confocal laser scanning microscopy
Confocal laser scanning microscopy (CLSM) is a method of light microscopy used to detect light emitted exclusively from a thin layer of a thick specimen. Fluorescent confocal microscopy is of special importance in biology and medicine, where it is used to analyse fluorescent signals from fixed and stained specimens, and from living cells and tissues. Optical principles that apply in confocal microscopy are identical to those used in the design of standard fluorescence microscopes. However, construction and operation of modern confocal microscopes introduces many important innovations related to methods of excitation, detection and interpretation of obtained images.
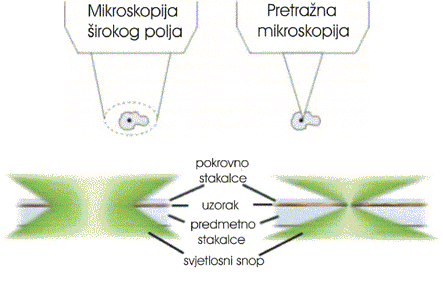 When contrasting
the similarities and differences between wide-field and
confocal microscopes, it is often useful to compare the character and geometry
of specimen illumination utilized for each of the techniques. Traditional wide-field
epi-fluorescence microscope objectives focus a wide cone of illumination over a
large volume of the specimen, which is uniformly and simultaneously illuminated
(as illustrated in Figure 1, left). A majority of the
fluorescence emission directed back towards the microscope is gathered by the
objective (depending upon the numerical aperture) and projected into the
eyepieces or detector. The result is a significant amount of signal due to
emitted background light and autofluorescence originating from areas above and
below the focal plane, which seriously reduces resolution and image contrast.
The laser illumination source in confocal microscopy is first expanded to fill
the objective rear aperture, and then focused by the lens system to a very small
spot at the focal plane (Figure 1, right). The size of
the illumination point ranges from approximately 0.25 to 0.8 micrometers in
diameter (depending upon the objective numerical aperture) and 0.5 to 1.5
micrometers deep at the brightest intensity. Confocal spot size is determined by
the microscope design, wavelength of incident laser light, objective
characteristics, scanning unit settings, and the specimen. Presented in Figure
1 is a comparison between the typical illumination cones
of a wide-field (Figure 1, left)
and point scanning confocal (Figure 1, right) microscope
at the same numerical aperture. The entire depth of the specimen over a wide
area is illuminated by the widefield microscope, while the sample is scanned
with a finely focused spot of illumination that is centered in the focal plane
in the confocal microscope.
When contrasting
the similarities and differences between wide-field and
confocal microscopes, it is often useful to compare the character and geometry
of specimen illumination utilized for each of the techniques. Traditional wide-field
epi-fluorescence microscope objectives focus a wide cone of illumination over a
large volume of the specimen, which is uniformly and simultaneously illuminated
(as illustrated in Figure 1, left). A majority of the
fluorescence emission directed back towards the microscope is gathered by the
objective (depending upon the numerical aperture) and projected into the
eyepieces or detector. The result is a significant amount of signal due to
emitted background light and autofluorescence originating from areas above and
below the focal plane, which seriously reduces resolution and image contrast.
The laser illumination source in confocal microscopy is first expanded to fill
the objective rear aperture, and then focused by the lens system to a very small
spot at the focal plane (Figure 1, right). The size of
the illumination point ranges from approximately 0.25 to 0.8 micrometers in
diameter (depending upon the objective numerical aperture) and 0.5 to 1.5
micrometers deep at the brightest intensity. Confocal spot size is determined by
the microscope design, wavelength of incident laser light, objective
characteristics, scanning unit settings, and the specimen. Presented in Figure
1 is a comparison between the typical illumination cones
of a wide-field (Figure 1, left)
and point scanning confocal (Figure 1, right) microscope
at the same numerical aperture. The entire depth of the specimen over a wide
area is illuminated by the widefield microscope, while the sample is scanned
with a finely focused spot of illumination that is centered in the focal plane
in the confocal microscope.
The confocal principle
in epi-fluorescence laser scanning microscopy is diagrammatically presented in
Figure 2. Coherent light emitted by the laser system (excitation source) passes
through a pinhole aperture that is situated in a conjugate plane (confocal) with
a scanning point on the specimen and a second pinhole aperture positioned in
front of the detector (a photomultiplier tube). As the laser is reflected by a
dichromatic mirror and scanned across the specimen in a defined focal plane,
secondary fluorescence emitted from points on the specimen (in the same focal
plane) pass back through the dichromatic mirror and are focused as a confocal
point at the detector pinhole aperture.
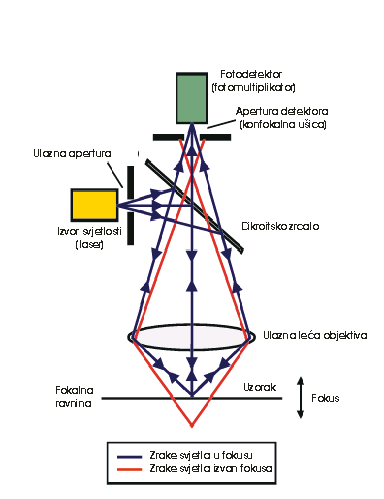 The
significant amount of fluorescence emission that occurs at points above and
below the objective focal plane is not confocal with the pinhole and forms
extended Airy disks in the aperture plane. Because only a small fraction of the
out-of-focus fluorescence emission is delivered through the pinhole aperture,
most of this extraneous light is not detected by the photomultiplier and does
not contribute to the resulting image. The dichromatic mirror, barrier filter,
and excitation filter perform similar functions to identical components in a
widefield epi-fluorescence microscope. Refocusing the objective in a confocal
microscope shifts the excitation and emission points on a specimen to a new
plane that becomes confocal with the pinhole apertures of the light source and
detector.
The
significant amount of fluorescence emission that occurs at points above and
below the objective focal plane is not confocal with the pinhole and forms
extended Airy disks in the aperture plane. Because only a small fraction of the
out-of-focus fluorescence emission is delivered through the pinhole aperture,
most of this extraneous light is not detected by the photomultiplier and does
not contribute to the resulting image. The dichromatic mirror, barrier filter,
and excitation filter perform similar functions to identical components in a
widefield epi-fluorescence microscope. Refocusing the objective in a confocal
microscope shifts the excitation and emission points on a specimen to a new
plane that becomes confocal with the pinhole apertures of the light source and
detector.
U praktičnoj izvedbi konfokalnih
mikroskopa najvažniji dijelovi su sklopovi za pretraživanje, tj. vođenje pobudne
i emitirane svjetlosti, tzv. skeneri, te detektori. Kao skeneri se najčešće
koriste sustavi elektronički vrlo precizno upravljanih zrcala, a kao detektori
brojači fotona, uglavnom fotomultiplikatori. Teoretska moć razlučivanja
konfokalnog mikroskopa određena je, kao i u klasičnom slučaju, kutom otvora
ulazne leće objektiva, tj. njegovom numeričkom aperturom, te valnom duljinom
fluorescentne svjetlosti. Maksimalno povećanje, međutim, nije određeno faktorom
optičkog povećanja objektiva. Ono je određeno isključivo tzv. zoom faktorom koji
se može kontinuirano podešavati i odražava veličinu rasterkog polja skenera.
Moguće je također odabrati i broj digitalnih elemenata rezultirajuće slike, tzv.
pixela, odabirom veličine, odnosno formata slike. Preporuča se ove parametre
uskladiti tako da je veličina pixela dva do tri puta manja od najmanje linearne
dimenzije koju je moguće razlučiti uz date optičke uvjete.
Iako je
potrebno uzeti u obzir moć prostornog razlučivanja konfokalnog mikroskopa, važno
je imati na umu da je vrijeme koje svaki pojedini točkasti element slike biva
osvjetljen vrlo kratko i u pravilu iznosi samo nekoliko mikrosekundi. U tom
periodu detektor je u stanju registrirati tek nekoliko desetaka emitiranih
fotona, tako da statistička svojstva brojanja fotona igraju važnu ulogu u
određivanju odnosa signala i šuma u slici. Uz određenu veličinu slike i zoom
faktor, odnos signal/šum moguće je poboljšati smanjenjem brzine skeniranja kao i
različitim metodama usrednjavanja (averaging). Treba imati na umu da se svim tim
metodama produžava vrijeme osvjetljenja svakog pojedinog pixela, a samim tim i
vrijeme potrebno za snimanje čitave slike. To je od posebne važnosti u slučaju
snimanja brzih procesa u živim stanicama. Osim takvih dinamičkih ograničenja,
vrijeme koje laserska zraka provede na određenoj točki uzorka (pixel dwell time)
potrebno je ograničiti i zbog efekata fotoizbjeljivanja i, u slučaju živih
stanica, fototoksičnosti.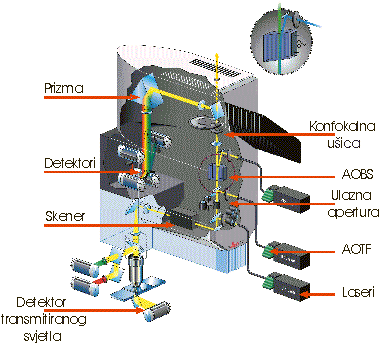
In our laboratory we are using the Leica
TCS SP2 AOBS confocal microscope (Fig. 3).
This instrument has
two characteristic features. Instead of
usual dichroic mirrors known from fluorescence microscopy,
this instrument utilizes an Acusto-Optical
Beam Splitter (AOBS). Furthermore,
instead of standard emission
filters, the total light emitted from the
sample is diffracted on a prism into a wavelength spectrum, which is further
divided to selected spectral regions by an array of apertures and mirrors. More
details about this instrument can be found on the
web page of the Haartman
Institute at the
University of Helsinki.