Time resolved photoelectron spectroscopy as a probe for ultrafast dynamics
With the newly available light sources in the XUV frequency domain, time resolved photoelectron spectroscopy (TRPES) has emerged as a leading technique for monitoring non-adiabatic excited state dynamics. To fully exploit the potential of this technique, new theoretical methods for calculating ultrafast spectroscopic observables need to be developed.
Research team
Nađa Došlić
Piero Decleva
Aurora Ponzi
Marin Sapunar
Sermsiri Chaiwongwattana
Zlatko BrkljaДЌa
Zoran Miličević
Ivan Ljubić
David Smith
Goran Zgrablić
List of publications:
[1]
M. Sapunar, A. Ponzi, S. Chaiwongwattana, M. Mališ, A. Prlj, P. Decleva and N. Došlić,
Timescales of N–H bond dissociation in pyrrole: a nonadiabatic dynamics study,
Phys. Chem. Chem. Phys., 2015, 17, 19012-19020.
[2]
V. M. Silkin, P. Lazić, N. Došlić, H. Petek and B. Gumhalter,
Ultrafast electronic response of Ag(111) and Cu(111) surfaces: From early excitonic transients to saturated image potential,
Phys. Rev. B, 2015, 92, 155405.
[3]
S. Chaiwongwattana, M. Sapunar, A. Ponzi, P. Decleva and N. Došlić,
Exploration of Excited State Deactivation Pathways of Adenine Monohydrates,
J. Phys. Chem. A, 2015, DOI: 10.1021/acs.jpca.5b07496
[4]
J. Thisuwan, S. Chaiwongwattana, K. Sagarik and N. Došlić
Photochemical Deactivation Pathways of Microsolvated Hydroxylamine, Submitted Manuscript
[5]
A. Ponzi, M. Sapunar, C. Angeli, R. Cimiraglia, N. Došlić and P. Decleva
Photoionization of furan from the ground and excited electronic states, Submitted Manuscript
Nonadiabatic dynamics of pyrrole
We investigated the excitation wavelength dependent photodynamics of pyrrole, a prototypical molecule displaying πσ*
mediated excited state deactivation. Nonadiabatic trajectory-surface-hopping dynamics simulations based on the TDDFT and ADC(2) methods were employed.
It was confirmed that upon excitation directly to the S1(πσ*) state, the deactivation is driven by tunnelling through a barrier of ΔE =
1780 cm-1. When the excitation energy is enough to overcome the barrier (236-250 nm), direct dissociation of the N–H bond takes place with
a time constant of 28 fs. Excitation in the 198-202 nm range to the bright B2 state returns a time constant for N–H fission of 48 fs at the
B3LYP/def2-TZVPD level, in perfect agreement with the experiment. For the same wavelength range, the ADC(2)/aug-cc-pVDZ constant is more than three
times larger. The discrepancy of the methods in the description of the relaxation from the B2 state was explained in terms of the basis set dependence
of the mixing between valence and Rydberg states. More.
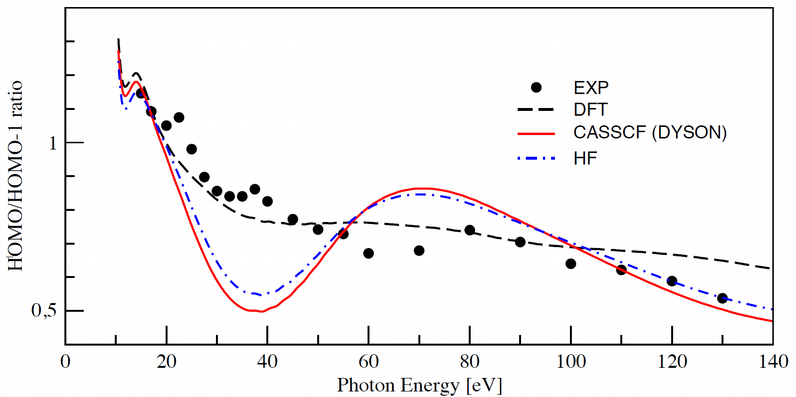
Photoionization of furan
In this work we focus on the photoionization of furan, a prototypical photochemical system, both from the ground and lowest-lying excited electronic
states. The Dyson orbitals (implemented for TDDFT, ADC(2) and CASSCF methods) approach is used together with an accurate description of the molecular
continuum obtained using a multicentric basis of B-spline functions to evaluate cross sections and asymmetry parameters. Apart from the ground state
minimum energy structure we considered a bent structure that is typically encountered along the relaxation pathway of furan after optical excitation
to the bright B2 state. The multicentric B-spline approach has proven to be a very accurate way to describe single-electron ionization problems and is
used for the first time in the context of molecular photoionization from excited electronic states in order to provide benchmark results against which
more approximate, but also more efficient, methods can be compared.
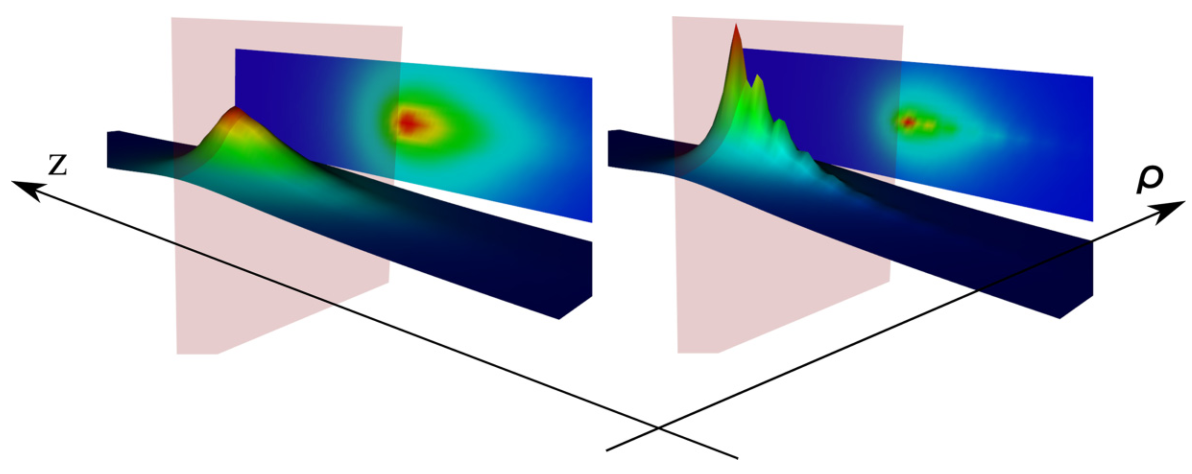
Ultrafast electronic response
We investigated the evolution of attosecond to femtosecond screening and emergent potentials that govern the dynamics and energetics of electrons and
holes excited in the various stages of multiphoton photoemission processes and control the photoelectron yield in recently reported experiments. The
study was focused on the dynamical screening of holes created in preexistent quasi-two-dimensional Shockley state bands on Ag(111) and Cu(111) surfaces
and of electrons excited to the intermediate and emerging screened states. The obtained results enable to establish a consistent picture of transient
electron dynamics at Ag(111) and Cu(111) surfaces that are becoming accessible by the time-, energy-, and momentum-resolved pump-probe multiphoton
photoelectron spectroscopies. More.
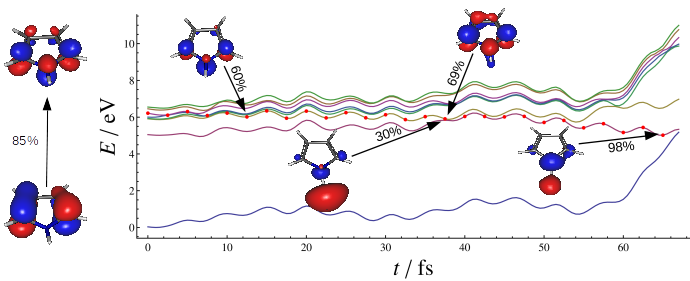
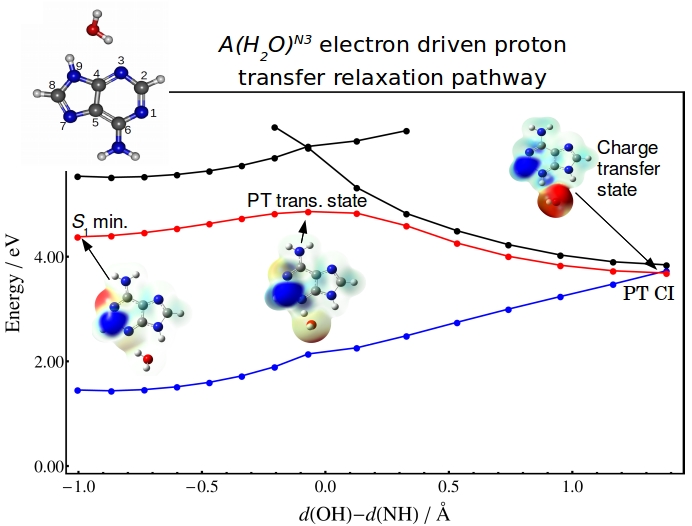
Scrutinizing the role of water in deactivation of biomolecules
It was predicted that binding of a single water molecule has a dramatic effect on the excited state lifetime of a number of biological chromophores.
In our work, we have attempted to understand the sub-100 fs excitation lifetime in adenine monohydrates by applying nonadiabatic dynamics and reaction
path calculations. Lifetimes computed through dynamics simulations at the ADC(2) level were found to be significantly longer that the observed one.
By comparing the reaction pathways of several excited state deactivation processes in adenine and adenine monohydrates, we have shown that
electron-driven proton transfer from water to nitrogen atom N3 of the adenine ring may be the process responsible for the observed ultrafast decay.
More.
![]()
![]()
The project was funded by The Unity Through Knowledge Fund (UKF).
Duration of the project: 24 months
Financing: 1 397 365.00 HRK